近年来,随着检验技术的日新月异,人类基因体学研究与分子检测技术迅速发展,许多新医学检验项目从实验室中被研发出来至临床应用,实验室自建检测(Laboratory Developed Tests,LDTs)成为快速成长的服务项目,更被视为实现预防医学与精准医疗的重要推手。而随着LDTs的大幅增加及商业化发展,如何在创新发展与监管之间拿捏,亦成为各国监理机关的重要课题。
图一、台湾地区资策会科法所法律研究员吕政谚在《精准健康法制研讨会》中分享LDTs法规的最新趋势。

吴碧娥/摄影
LDTs是指在医学实验室中自行研究发展、制造、验证且仅供该实验室使用的检测方法。LDTs 并非近年才出现的新名词,然而过去医学检验实验室发展的LDTs,多数使用手工检测方式搭配人为判读来进行各项诊断,多为需求量小、测试简单、且非商业用途性质,因此各国过去皆未将LDTs 纳入强制管理。但因近年使用LDTs检测作为辅助诊断与治疗决策的临床需求剧增,为确保于临床实验室自行研发检验技术时,具有充分的风险管控,且能达到所宣称的功效,各国卫生主管单位倾向建立适度的规范,对LDTs进行监管[1]。
精准医疗已是全球医疗、健康产业发展的重要趋势,台湾除了积极推动精准健康产业,也针对实验室开发检测制定相关法规。台湾地区资策会科法所法律研究员吕政谚指出,根据2021年修正的「特定医疗技术检查检验医疗仪器施行或使用管理办法」(特管法),已将LDTs纳入管理,实验室需取得质量认证,只是目前施行以来尚有优化空间。台湾精准医疗产业协会所提出的《2023精准健康产业政策白皮书》,为发展台湾精准医疗提出五大建言,其中一项就是优化医疗机构申请LDTs的申请程序与管理,以提高精准健康检测量能、速度,并进一步降低检测成本。
美国修法尚未通过
LDTs在美国是由联邦医疗保险和补助服务中心(The Centers for Medicare and Medicaid Services,CMS)进行实验室认证,随着LDTs的数量及类型大幅增加、发展越趋商业化,美国食品药品管理局(Food and Drug Administration,FDA) 在2014年发布LDTs监管架构指引草案,以风险基础方法对LDTs加以监管和进行分级。除了应向FDA通知或注册LDTs,若进行重大改变也必须额外通知FDA,结果引发各界抨击。FDA另于2017年发布LDTs讨论文件,尚未执行此监管架构。
2018年,美国国会议员提出由FDA监管的《评估精准且先进的体外临床检验技术发展法》(Verifying Accurate,Leading-edge /VCT Development Act, VALID Act)草案、参议院议员另提出由CMS监管之《评估美国验室之创新检测法》(Verified Innovative Testing in American Laboratories Act,VITAL Act)草案,虽然在116届和117届美国国会都曾提出过法案,至今皆仍未通过。
VALID Act将LDTs列为「体外临床检测」(In vitro clinical tests,IVCTs),认为应有独立的监管架构,必须依照对病人和公众危害严重程度,分为高、中、低风险。除了要求临床确效报告使用真实世界数据,还要制定FDA咨询机制并举行公听会。吕政谚指出,VALID Act比《临床实验室改进修正案》(CLIA)更为严格,相关规定涵盖管理职责、质量稽核要求、人员、设计管理、文件管理等各方面要求,反对者认为VALID Act是在阻碍创新,加上FDA缺足够的资源履行这些义务,不可预测的监管流程将会造成成本增加。2020年美国卫生及公共服务部(United States Department of Health and Human Services,HHS)也禁止FDA针对LDTs进行上市前审查。不过,无论VALID Act是否通过,FDA仍可能以其他方式监管LDTs。
图二、美国LDTs监管架构
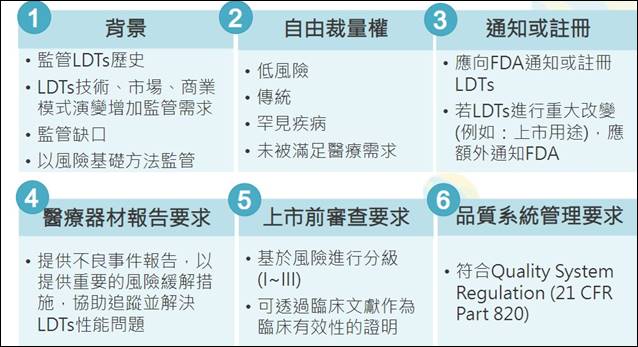
图片来源:2023/8/16,台湾地区资策会科技法律研究所《精准健康法制研讨会》,吕政谚简报数据
欧盟全面实施IVDR
欧盟将LDTs定义为「医疗机构(Health institutions)在内部自行制造、变更和使用自行研发检测,并未以工业规模生产,来解决目标病人群体的需求,而病人无法在市场上自行购买相同效能的检测」。欧盟已于2022年5月26日全面实施「体外诊断医疗器材法规」(IVDR),取代原有的《体外诊断医疗器材指令》(IVDD),由欧盟执行委员会(EC)、医疗器材协调小组执行,超过八成的产品需要透过第三方机构验证。
IVDR将产品依风险分成A~D级,需第三机构验证的产品从20%大幅提高至80%。原先取得IVDD验证的产品,仍须透过第三方机构进行IVDR验证,不仅技术文件及临床数据要求增加,并强化产品可追溯性,要求透过欧盟医疗器材数据库(EU medical devices database,EUDAMED)向大众公开安全与性能摘要。此外,除了上市后监督,每五年还会对产品进行一次无预警稽核。考虑到目前第三方机构验证能量不足,欧盟已依医材风险等级不同提供延长过渡期。
图三、欧盟LDTs监管架构
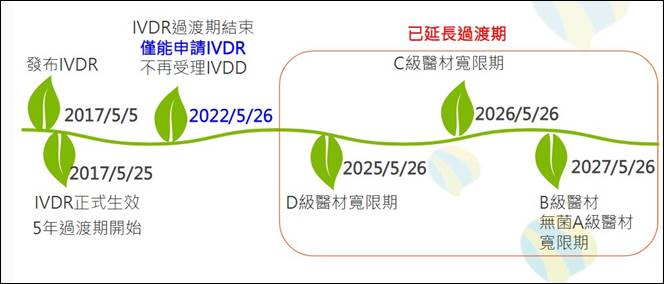
图片来源:2023/8/16,台湾地区资策会科技法律研究所《精准健康法制研讨会》,吕政谚简报数据
欧盟执行委员会在今年7月底发布调查结果,共有1551件IVDD产品待申请IVDR,但2023年仅950件申请、331件取得IVDR认证,相较2022年申请数量上升15.6%。吕政谚指出,六成以上产品取得IVDR认证需要18个月时间,若有需要应及早进行IVDR送审,以免影响后续产品上市计划。
韩国正面表列LDTs可检测项目
韩国食品药物安全部(Ministry of Food and Drug Safety,MFDS)在2019年修订《体外诊断医疗器材法》,将LDTs纳入监管并于2020年5月1日开始实施,将产品依风险分1~4级,类似于欧盟IVDR风险分级方式,并正面表列基因检测机构可进行检测的项目。
根据韩国医疗法第3条规定,医疗机构、生物伦理及生物安全法第49条规定之基因检测机构,应备有自行设计、规划体诊断检测系统,而且仅能在其临床实验室内部使用;而韩国临床实验室应取得监管机关食品药物安全部自行发展的认证制度,认证重点包括:临床实验室质量管理系统、体外诊断医疗器材的性能,并由MFDS评估检测专业人员的资格及经验。
此外,韩国食品药物安全部在2016年修订《生物伦理及生物安全法》,将消费者基因检测(direct-to-consumer genetic testing,DTC)纳入监管,最初先是允许12项DTC基因检测,2020年进一步扩增至70项。吕政谚指出,在韩国可以由医疗机构以外的基因检测机构采集、检测、分析检测结果,并将测试结果直接提供给消费者,适用范畴仅限定健康领域,或落发、皮肤老化等外观特征,若涉及疾病风险,仍应由医疗机构进行检测。
日本尚未设立专法
日本则未设立LDTs专法,将LDTs称为「自家调整检查法」,医疗用途的基因检测仅能在医疗机构中执行。2017年,日本修正及《医疗法》及《临床检验师法》,依检验行为及检验场所分为医疗机构自行检查、委托其他医疗机构检查、委托医事检查所检查,主管机关为厚生劳动省(Ministry of Health, Labour and Welfare,MHLW)和医药品医疗器材综合机构(Pharmaceuticals and Medical Devices Agency,PMDA)。日本目前已核准3项用于癌症基因的下一代测序技术(NGS)检测上市,由国民健康保险部分负担,只能在获得MHLW核可的医院下进行检测。
吕政谚建议,台湾可参考国际上对于LDTs分类方式,制定相对应的监管机制,也可参考美、欧、韩的做法,依照对病人或公众危害严重程度进行风险分类,透过生技公司与医疗机构的协力合作,一同推动台湾LDTs产业的发展。
数据源:
- 2023/8/16,台湾地区资策会科技法律研究所《精准健康法制研讨会》,吕政谚简报。
备注:
好消息~北美智权报有微信公众号了!
《北美智权报》内容涵盖世界各国的知识产权新闻、重要的侵权诉讼案例分析、法规解析,以及产业与技术新知等等。
立即关注北美智权微信公众号→ NAIP_IPServices
~欢迎读者分享与转发~ |
 |
|
| 作者: |
吴碧娥 |
| 现任: |
北美智权报主编 |
| 学历: |
(台湾)政治大学新闻研究所 |
| 经历: |
北美智权报资深编辑
骅讯电子总经理室特助
经济日报财经组记者
东森购物总经理室经营企划 |
|
|
|


