全球20种最畅销药物中,高达7种是治疗性单株抗体,这些抗体药可以治疗多种疾病,包括类风湿性关节炎、自身免疫性疾病和多种癌症。抗体药物每年为制药公司带来了数十亿美元市场,因此使激励药厂致力于开发生物药物。

图片来源 : shutterstock、达志影像
随着生物药物市场竞争的激增,生物制药公司为确保竞争力,也尽可能以专利保护所开发的新药。另一方面,生物药品市场因为竞争者的加入,许多新的专利争议也随之而生,若干生物药品专利有效性判断原则因此而所调整。由于抗体开发技术之进展,有关专利说明书记载的内容,是否可支持专利请求项之判断标准,也有趋严之趋势。
抗体药市场
伴随着生物科技的发展与临床使用需求,治疗性单株抗体跃居全球最畅销药品,用于治疗多种疾病,包括类风湿性关节炎、自身免疫疾病和多种癌症。治疗性单株抗体于医疗市场所带来的商业利益更加显著,例如pembrolizumab为Merck & Co.开发之PD1阻断剂,于2019年的收入超过110亿美元,至2025年可能高达240亿美元。Bristol Myers Squibb的PD1阻断剂nivolumab于2019年的收入为80亿美元,预计很快将突破100亿美元的门坎。
依据Cortellis的数据显示,2019年抗体占销售额前20名的治疗药物中的9种,累计收益为750亿美元。由此可见与抗体相关的发明价值非常可观,尤其开发抗体疗法需要大量资源与时间,因此,制定强有力的专利策略以保护投资、防止逆向工程,尽可能减少治疗性抗体设计上的变更至关重要,原厂药与生物相似药品于市场高度竞争的情况屡见不鲜,衍生的专利诉讼挑战日益严峻。
单株抗体发展现况
抗体亦称为免疫球蛋白(Immunoglobulin,Ig),形状类似Y型分子,由4条多肽链组成,含有两条重链与两条轻链,彼此透过双硫键连接。目前单株抗体形式如图1所示:经典(a)、抗体药物结合物(b):由合成之连接分子与小分子药物键结而组成、双特异性(c):两种特异性抗原结合位,可同时作用于两种标靶,通常一部分结合至肿瘤细胞抗原,另一部分结合至T细胞上,另一种形式为两个抗原结合位,可结合至两种目标抗原。还有片段型式(d)包括抗原结合片段(Fab)、单链可变区(scFv)构建体和奈米抗体。
图1:各类抗体型式示意图
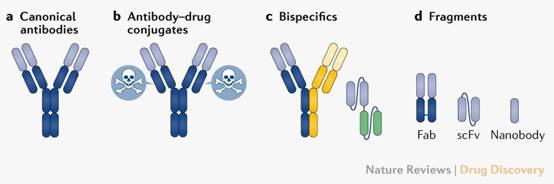
数据源:Nature Reviews Drug Discovery, volume 20, July 2021, 491-495
1975年科学家乔治.科勒(Georges Köhler)和塞萨尔•米尔斯坦(C塞萨尔•米尔斯坦öhler)研究杂交瘤技术,利用抗原接种小鼠,提取脾细胞,将B细胞与骨髓瘤细胞(myeloma cells)成功地合成融合瘤细胞,培养出可分泌抗体的B细胞,开启单株抗体(mAb, monoclonal antibody)研究的先河,使得大量获得纯mAb成为可能,极大地增强了基础研究和临床应用的潜力,此项重要发明于1984年获得诺贝尔奖(单株抗体生产示意图如图2)。
图2:单株抗体生产示意图
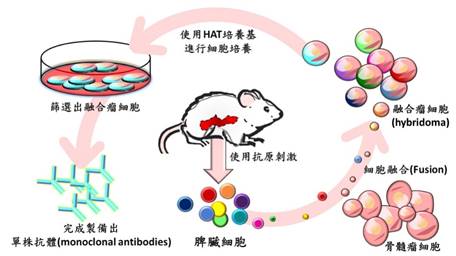
参考数据:https://www2.mrc-lmb.cam.ac.uk/achievements/lmb-nobel-prizes/1984-cesar-milstein-georges-kohler/
1979年,Schlossman与合作者已经确定了三种针对独特T细胞抗原的单株抗体,其中一个被称为OKT3。1981年,他们在临床中测试OKT3作为预防移植排斥的免疫抑制剂。1986年,OKT3更名为muromonab-CD3,获得US FDA批准,成为第一个上市应用于全美医疗市场之治疗性单株抗体。直到8年后的1994年,FDA才批准了第二种治疗性单株抗体产品。
由于细胞分子生物技术的发展日益成熟,许多复杂且耗时的技术被新颖的生物技术取代,使大量制造治疗性单株抗体变为可行,更加速治疗性单株抗体的发展,自2006年起,抗体药物,平均每年大约有10个抗体药物获得US FDA批准上市,提供病患使用。2015年US FDA批准了第50个抗体,比第一个抗体的核准晚了29年。2021年4月葛兰素史克(GSK)公司的PD1 抑制剂dostarlimab,获得US FDA批准,US FDA核准上市之治疗性抗体已达100项,仅用了6年时间,前10大治疗性抗体针对的标靶目标统计如表1,分属13种治疗领域,其中用于癌症治疗)占41%、血液疾病与皮肤疾病各占9%、感染性疾病占7%、风湿免疫疾病占6%,其他领域28%。
表1:前100个mAb之标靶目标统计
标靶目标 |
单株抗体数目 |
PD1/PDL1 |
7 |
CD20 |
6 |
TNF |
4 |
HER2 |
4 |
CGRP/CGRPR |
4 |
VEGF/VEGFR |
4 |
IL-6/IL-6R |
4 |
IL23 p19 |
3 |
EGFR |
3 |
CD19 |
3 |
数据源:The Antibody Society, Nature Reviews Drug Discovery.
单株抗体之生物相似性与审查考虑
药物化学家可能要花费数年时间才能找到对特定目标具有活性的小分子,而抗体的发现仅需要几个月。根据Reichert对2005年至2014年间进入临床的569种抗体的分析,从自临床试验第一期开始至核准上市之总体成功率为22%,抗体药品在临床试验中成功的可能性是小分子药品的2-3倍。
艾伯维(AbbVie)类风湿生物制剂Adalimumab(Humira),系以TNF-α为标靶之单株抗体,适用于患有中度至重度类风湿性关节炎,并且曾经对一种或超过一种的DMARDs药物有不适当反应的成人病患,可减轻症状与征兆。2002年核准上市,仅2019年就为药厂带来近200亿美元收入,为目前药界最畅销的药品。娇生药厂的infliximab(Remicade)对TNF-α有很高的亲和力,可以阻碍TNF-α和其受体结合,亦可殺死表现TNF-α受体的细胞,2015年年销售额达到100亿美元巅峰,虽比Humira早4年核准上市,该单株抗体2019年仍超过50亿美元。
由此可见,单株抗体药物具备较一般药品高的研发成功率,上市后更可为药厂带来庞大商机,进一步带动各大药厂投入治疗性单株药品抗体药品或其生物相似药的开发,涵盖这些抗体药品的专利价值达数10亿美元,如何保护研发的重要技术与获得广泛的专利保护成为制药公司的巨大挑战。
单株抗体要应用于临床使用,必须考虑抗体应具有较低免疫原性(immunogenicity)、高度专一性(specificity)、抗原结合能力(affinity)、诱发内化作用等因素,相关法规可参考中华药典(4029)单株抗体制剂、中华药典(4031)基因工程制剂、药品查验登记审查准则-基因工程药品之查验登记,以及生物相似性单株抗体药品查验登记基准。
另一方面,由于生物药品结构较化学药品庞大且复杂,具有制程专一性,两种生物药品是否具备相似之疗效与安全性,必须就化学制造与管制(CMC)、药理毒理、药动药效及临床进行比较,尤须着重于比较两者间是否相似,其临床试验应以探讨试验药品与参考药品之临床疗效是否具显著差异为主要设计,在受试者人数估算方面则以须足够鉴别试验药品与参考药品之不良反应,并提供免疫原性与疗效、安全性之关联数据为考虑重点[1]。生物相似性药品获准上市后,主管机关亦会视审查情形,请药厂提供上市后药品安全监测计划,持续监控药品临床使用之安全性。
抗体药物技术开发与专利保护范围之演变
对于药厂而言,抗体药物之开发与其他药物开发相同,需尽早申请专利以保护潜在创新。科学家开始发展治疗抗体技术之初,当时抗体相关专利申请,仅揭露少部分的抗体信息就可以取得专利保护。但随着治疗性抗体技术逐渐成熟,制药公司需要提供更多的数据,才可以获得较大的专利保护范围。特别是,在美国发明法案(AIA)施行之后,缺乏必要的数据将无法证明该发明可以实施,最终导致无法取得专利保护。
根据研究指出,90年代抗体专利申请案,许多仅公开paratope(抗原结合位,指与抗原或蛋白结合的抗体部分)或epitope(抗原表位,与抗原结合的目标蛋白部分),足以取得专利[2]。当时,专利申请人无需揭露与该抗原结合的实际抗体,因此专利请求项仅需记载「一种与抗原结合的抗体(记载接合区域)」,如此一来,由于抗体专利请求项,仅有与特定抗原结合,因此可使专利申请人获得大范围的抗体专利之保护[3]。而公司取得抗原表位或结合位后之较大专利保护范围后,其后仍可再藉由揭露抗体真正序列,以获得更小的抗体专利。这些专利权利的要求,构成了最早抗体专利的基础。
随着早期抗体专利陆续到期,生物制药公司继续开发第二代抗体。但是,想要取得早期抗体专利的广泛保护范围,变得越来越困难。首先,新的专利需克服可预期和显而易见的问题,申请人必须证明新抗体和旧抗体之间有足够的差异。此外,对于一个抗体属(genus)的专利保护,近年来美国联邦法院对于专利说明书的书面记载之要求,有更严格的条件,因此过去判决所形成「抗体例外」原则,逐渐不被采用。在新的司法实务的影响下,新的专利说明书之书面记载条件,要求需要保护特定属的抗体,应提供足够代表性数量的抗体和/或提供更详细的表位讯息。
1.抗体例外原则
1997年在Regents of the University of California v Eli Lilly[4]案乙案,本案虽然与抗体没有直接关系,但判决内容与抗体专利申请面临的问题相关[5]。本案法院确认一项说明书揭露原则,即特定属的生物序列要获得专利之保护,必须具备足够属于该属实例的代表案例。本案因原告加州大学所有之专利,其涉及DNA基因重组技术,而该技术可产生人胰岛素。一般情况,健康人通过对前胰岛素原(PPI,preproinsulin)进行末端酶促裂解来产生胰岛素原(PI, proinsulin),即一个单一的氨基酸链,从而在体内产生胰岛素。该专利的申请,则是基于对大鼠中发现的PI和PPI cDNA序列的测定。专利说明书提到cDNA是本发明的一部分,但说明书仅提供了生产人胰岛素cDNA的一般方法,并提供对人胰岛素cDNA的描述。原告起诉被告侵权,被告辩称专利无效。地区法院审理时,以说明书没有提供足够的cDNA书面描述,而裁定该专利无效,导致原告败诉。原告因而提出上诉。
联邦巡回上诉法院(CAFC)判决中指出,一家公司要求保护一个属的生物序列(DNA序列),说明书必须揭露具有代表性数量的该属实例。然而根据Noelle v. Lederman案,这一书面揭露要求的先例,并未适用于抗体专利。[6]美国专利商标局(USPTO)而后适用Noelle v. Lederma案之法理,修正审查基准,而衍生有「抗体例外」原则,即允许专利发明人在不描述抗体真正情况下仍能可以保护该抗体。此一原则,根基于设定抗体-抗原关系有如锁和钥匙,因此只要发现一个新的抗原,并能充分表现其结构,那么创造与其结合的抗体,就是一个常规实验的问题。因此,在申请抗体专利的情况下,公司可以透过揭露抗原表位,与该抗原结合的分离抗体,无需披露此类抗体就可以获得保护。
2.抗体例外原则适用之限缩
2011年Centocor Ortho Biotech, Inc. v. Abbott Laboratories[7]乙案,则开始挑战所谓抗体原则之例外。本案涉及人类肿瘤坏死因子α(Human necrosis factor α 以下简称“TNF-α”)的抗体开发。当时,Centocor和Abbott都试图开发具有高亲和力、具备中和活性和低免疫原性(immunogenicity )的中和TNF-α因子的治疗抗体(therapeutic TNF- antibodies),但二者采用不同的策略来开发抗TNF-α抗体。本案争议专利,为Centocor公司持有之美国专利,专利号7,070,775 。775专利为Centocor开发之抗体相关技术。Centocor采用一种既有技术,藉由修改小鼠抗体,将抗体的小鼠恒定区与人类恒定区交换,以降低抗体的免疫原性,从而产生嵌合抗体,因而开发出TNF-α因子的小鼠抗体(“A2小鼠抗体”),该抗体对于人类TNF-α因子具有高亲和力和中和活性。与此同时,Abbott采用不同技术开发全人类抗体,其藉由筛选人类可变区噬菌体库(phage display library),而筛选并定位可与人类TNF-α因子结合之可变区,然后使用各种技术来提高所选可变区的结合亲和力。然后将所得可变区与人恒定区组合以产生全人抗体,1995年,Abbott藉由以上技术而开发出治疗性抗体Humira®。
Abbott 于1996年提出专利申请,该专利揭露对TNF-α的高亲和力、具有中和力之全人抗体,即Humira®抗体,并于2000年获得专利,专利号为 6,090,382。在上诉审理中,其中的关键问题涉及'775专利,是否为针对保护人类可变区提供了充分的书面描述。如上所述,Centocor于2002年首次寻求对人可变区和全人抗体的专利权利范围之保护。但当时,Abbott已经发现了一种具有高亲和力和中和活性的TNF-α全人抗体,并为其创新申请专利。因此,于诉讼中,Centocor就对人可变区和全人抗体的专利权利范围之保护,除非可主张较早申请案的优先权日期,否则并无法控告Abbott侵权。由于Abbott的专利申请系于1996年提出的,于诉讼中Centocor需要胜诉,仅能依赖于其于1994年CIP申请的优先权且其所主张的权利必须得到1994年CIP申请的充分书面描述的支持。
首先,上诉法院认为,提出如何制造一种类型的抗体(即嵌合)的描述,并不支持制造不同类型(即人类)抗体的请求项。因此,法院认为Centocor请求项的范围,比其说明书记载之内容要大。本案确立专利申请人不再可以透过简单地描述抗原,而有权获得与其结合的任何类型的抗体。如果需要保护特定类型之抗体,必须描述该特定类型之抗体。
3.专利范围需有充足数据支持
在Amgen v. Sanofi[8]案中,CAFC裁定,为了获得与特定抗原结合并发挥特定功能的一类抗体的广泛专利覆盖范围,公司必须披露足够数量以跨越所要求保护的属的代表性抗体,或建立抗体的功能与其说明书中的抗体属之间的明确关系。
本案专利涉及一种单株抗体,该抗体与目标抗原序列上的15个不同氨基酸残基中的一个或多个结合。该专利请求向,包括抗体何时阻止抗原与其目标结合的功能性用语。Amgen之专利申请,包含结合15个残基之一或其任意组合的抗体。但该专利案,仅公开了两种抗体,但有特异性结合数据和亲和力数据的支持。如果以Centocor的判断标准下,本案专利书面记载的条件应已符合,因为说明书已经对于抗体提出特异性结合数据和亲和力数据。然而,CAFC判决要保护一属的抗体,仅揭露二种抗体是不够的,法院要求专利说明书必须公开足以代表性数量之案例,才能满足书面说明。
又在本案中之争议专利并未揭露抗体结构的任何内容(例如氨基酸序列或3D结构)。因此,被告质疑与目标抗原结合之抗体专利,可能涵盖数百万种抗体,即使该领域技术之人,也无法制造和使用每一种抗体。一审法院认为,即使有许多能够结合的抗体的实施例,该领域技术人员也无法制造和使用能够进行这种结合的广泛类别(或「属」)的抗体。
CAFC认为制造和使用所需提供之实验数量,不仅需考虑专利公开的实施例数量,而且还包括权利的完整范围。法院引用Wands[9]中阐明的因素来分析,以说明是否提供足够实验数据以支持保护范围:(1)必要的实验数量,(2)提出的指导或指导的数量,(3)工作实例的存在与否,(4)性质(5)现有技术的状态,(6)本领域技术人员的相关技能,(7)本领域的可预测性或不可预测性,以及(8)权利要求的广度。
结语
有关各种抗体药开发之技术仍然在成长中,法院实务显示随着抗体开发技术成熟,以及对于抗体学了解越透彻,法院实务已朝向申请人须提供更多之实验数据已取得更大专利范围之保护。如果实验证据不足,未来在抗体专利申请可能面临专利无效的问题,然而,实验数据的生成需要时间。因此,如何提供足够数据与及早提出申请以保全专利申请优先权,考验实验室与专利申请人经验累积,与司法实务之发展。
备注:
- 参考数据:财团法人医药品查验中心当代医药法规月刊第92期
-
Karen Carroll & Sharad Bijanki, The Evolution of Antibody Patents, October 8, 2018, available at https://www.ipwatchdog.com/2018/10/08/evolution-antibody-patents/id=101971/.
-
同上。
-
Regents of Univ. of Cal. v. Eli Lilly & Co., 119 F.3d 1559, 1997 U.S. App. LEXIS 18221, 43 U.S.P.Q.2D (BNA) 1398 (Fed. Cir. July 22, 1997)
-
A description of a genus of cDNAs may be achieved by reciting the sequence of nucleotides the cDNA is comprised of. A description of a genus of cDNAs may be achieved by means of a representative number of cDNAs falling within the scope of the genus or a recitation of structural features common to the members of the genus, which features constitute a substantial portion of the genus
-
Noelle v. Lederman, 355 F.3d 1343 at 1349(Fed. Cir. 2004).
-
Centocor Ortho Biotech, Inc. v. Abbott Laboratories (Fed. Cir. 2011)
-
Amgen Inc. v. Sanofi, Aventisub LLC, 872 F.3d 1367
-
In re Wands, 858 F.2d 731, 737 (Fed. Cir. 1988)
好消息~北美智权报有微信公众号了!
《北美智权报》内容涵盖世界各国的知识产权新闻、重要的侵权诉讼案例分析、法规解析,以及产业与技术新知等等。
立即关注北美智权微信公众号→ NAIP_IPServices
~欢迎读者分享与转发~ |
 |
|
【本文只反映专家作者意见,不代表本报立场。】
| 作者: |
叶云卿 |
| 现任: |
北美智权(cn.naipo.com)外稿作家
台湾世新大学 知识产权研究所 副教授
台湾科技大学 专利所 兼任助理教授 |
| 学历: |
美国旧金山金门大学 法律博士(SJD)
美国华盛顿大学 法律硕士(LLM)
台湾政治大学 法律硕士
台湾大学环境工程所 工程硕士 |
| 经历: |
台湾科技大学专利所 助理教授
美国旧金山 Suzan See Law Office法务
美国硅谷 Vivian Lu Law Office法务
台湾建业律师联合事务所律师
台湾环宇律师事务所律师 |
| 产学合作计划: |
美国诉讼管理产学合作计划 |
| 代表著作: |
营业秘密刑事责任
中小企业知识产权管理制度建置
专利意见书在诉讼上之运用 |
| 證照: |
律师、台湾专利代理人、环境工程技师、仲裁人、ISO14000管理师 |
|
|
| 作者: |
张连成 |
| 现任: |
台湾阳明交通大学药物科学院药学系 兼任助理教授 |
| 学历: |
台湾阳明大学生物药学所博士 |
| 经历: |
TFDA 技正、副研究员、科长 |
|
|
|


